

1.4. 电子关联#
写于2025年3月4日
整理发布于2025年3月16日
之前整理了一下电子关联问题,也涉及到关联能,交换能等化学计算的重要问题。
在量子计算发展的早期,也就是当下时代,选择合适的化学问题来求解是重要的,强关联问题应当是这类问题的一个特征。
设非相对论电子哈密顿量为:
相同表述形式:
可以参加电子哈密顿量从BO近似的来源Born-Oppenheimer Approximation
一、基本近似#
任何量子化学方法均可分解为以下步骤:
基组截断 (Basis Set Truncation)#
备注
电子薛定谔方程的维度过于大,不能严格求解,必须通过近似方法。这一系列近似方法就导致了计算精准度的缺失。如图所示,使用有限的基组只能在一定范围内逼近波函数,但总会有误差。越大的基组误差越小,但对算力消耗大。选择合适的基组,在 HF 和 DFT 方法中都很重要。
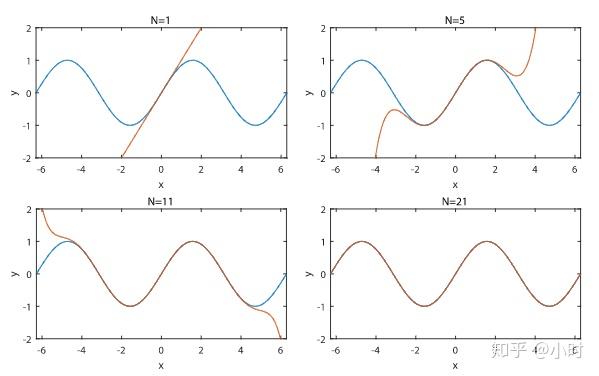
数学操作:将单电子轨道空间限制为有限维子空间 \( \text{Span}\{\chi_\mu\}_{\mu=1}^M \)
后果:引入基组误差(Basis Set Error),属于技术性近似。
例子: Cusp条件与基组问题
在非相对论量子力学中,当两个电子靠近时(\(r_{12} \to 0\)),波函数需满足Kato cusp条件:
这一条件确保电子间的库仑排斥发散(\(\frac{1}{r_{12}}\))被动能项精确抵消。然而,常用高斯型基组(如cc-pVDZ)在\(r_{12}=0\)处过于平滑,无法自然满足cusp,导致动态关联能被低估。

改进方法:
显式相关方法(如F12):在基组中引入显式依赖\(r_{12}\)的函数项。
基组外推:通过增大基组尺寸逼近完备基极限。
波函数反对称化(Antisymmetrization)#
备注
对电子这种费米子来说,严格的解应当只有反对称解。但是电子薛定谔方程的解存在对称解和它简并。因此,如果在求解时对称解和反对称解耦合,就会引起“交换能”。HF 在波函数形式上保证了反对称,而 DFT 没有,xc 泛函是包含交换能的。
HF,通过构造Slater行列式 \( |\Phi\rangle = \mathcal{A}(\phi_1 \phi_2 \cdots \phi_N) \), 这是完备的。
严格满足Pauli原理,无信息损失。
KS波函数:形式上为Slater行列式,满足反对称性,但仅用于构造电子密度,不直接参与能量计算。
交换关联势:通过泛函\(E_{xc}[\rho]\)隐式包含交换与关联效应,无显式反对称项。例如:
LDA泛函:\(E_{xc}^{\text{LDA}}[\rho] = \int \rho(\mathbf{r}) \epsilon_{xc}^{\text{hom}}(\rho(\mathbf{r})) d\mathbf{r}\),其中\(\epsilon_{xc}^{\text{hom}}\)为均匀电子气模型下的交换关联能密度。
杂化泛函(如B3LYP):混合HF交换与DFT交换,如\(E_x^{\text{B3LYP}} = a E_x^{\text{HF}} + (1-a) E_x^{\text{DFT}}\)。
单参考近似(Single-Reference Approximation)#
数学操作:假设真实波函数可展开为单个参考态 \( |\Phi_0\rangle \) 及其激发态的线性组合:
后果:引入关联能缺失(Correlation Energy Error)。
以下表述来自 Garnet Chan 的 lecture note.
备注
在 HF/DFT 类方法中,使用 SCF 的计算手段,我们首先得到的是一个基态。这个基态包含了单电子近似引起的问题。为了消除单电子近似的误差,HF 里使用激发态来补偿,而 DFT 使用 xc 泛函来补偿。Garnet Chan 用纠缠熵来表述,实际上是一种量子计算的视角。单电子近似只能表示直积态。


二、关联(correlation)#
从 HF 的视角出发,可以更清晰地看出关联之间的区别,我们假设通过 HF 已经选定了一个态做参考态,以此出发,通过它的激发态耦合出更低能量的态。
在给定基组中,如果所有激发态的线性组合被遍历,应该来说,这种搜索在这个子空间是完备的。不过这在计算上是不现实的。一般来讲,有两类在实际体系中常见的情况:
动态关联(Dynamic Correlation)#
备注
一般来说,动态关联指的是参考态有效,但高阶激发态较多,总体影响不小。这类情况常见于分子的平衡键长位置。可以通过 CC 方法快速收敛。
定义:在单参考态 \( |\Phi_0\rangle \) 的邻域内,可通过微扰展开或低阶激发态线性组合有效逼近的关联效应。
数学特征:
能量尺度:激发态与参考态能量差较大(\( \Delta E = \epsilon_a + \epsilon_b - \epsilon_i - \epsilon_j \gg |\langle \Phi_{ij}^{ab} | H | \Phi_0 \rangle| \))
波函数权重:主组态权重 \( |c_0|^2 \approx 1 \),激发态权重快速衰减(如 \( |c_i^a| \sim 10^{-2} \))
微扰收敛性:满足Brillouin定理(单激发态贡献为零),MP2、CCSD(T)等微扰方法收敛。
物理根源:电子瞬时位置涨落导致的库仑排斥修正(短程效应)。
静态关联(Static Correlation)#
备注
静态关联指的是参考态失效。也可以认为是 HF 的基态和单激发态能量非常接近,也可以说单电子轨道排布可能是不对的。真实基态往往是多个不同排布纠缠形成的,其能量显著低于 HF 基态。这种情况下,要考虑多参考态方法,例如 CAS.
定义:需要多个参考态非微扰叠加才能描述的关联效应,无法通过单参考激发态展开有效逼近。
数学特征:
能量简并:存在多个Slater行列式 \( \{|\Phi_I\rangle\} \) 满足 \( |\langle \Phi_I | H | \Phi_J \rangle| \sim |E_I - E_J| \)
波函数权重:多个组态权重相当(如 \( |c_1|^2 \approx |c_2|^2 \approx 0.5 \))
微扰发散:微扰展开失效(如H₂解离时MP2能量发散)。
物理根源:量子多体系统的非定域纠缠,如化学键断裂、过渡金属d电子强关联。(长程关联)
三、哈密顿量矩阵结构的可视化区分#
考虑组态相互作用(CI)的哈密顿矩阵:
动态关联主导:矩阵呈对角优势(对角元远大于非对角元),微扰展开有效。
\[ |H_{ii} - H_{jj}| \gg |H_{ij}| \quad (i \neq j) \]静态关联主导:存在子矩阵块,其中对角元接近,非对角元不可忽略。
\[\begin{split} \begin{pmatrix} H_{11} & H_{12} \\ H_{21} & H_{22} \end{pmatrix}, \quad |H_{11} - H_{22}| \sim |H_{12}| \end{split}\]
四、方法设计的数学指导#
1. 动态关联处理方法#
微扰理论(MP2, CCSD(T)):
利用Møller-Plesset拆分 \( H = H_0 + V \),其中 \( H_0 = \sum_i \epsilon_i a_i^\dagger a_i \)。
动态关联能通过二阶微扰项捕获:\[ E_{\text{corr}}^{(2)} = \sum_{i<j, a<b} \frac{|\langle \Phi_0 | V | \Phi_{ij}^{ab} \rangle|^2}{\epsilon_i + \epsilon_j - \epsilon_a - \epsilon_b} \]耦合簇(CC):
指数参数化 \( |\Psi\rangle = e^{T} |\Phi_0\rangle \),其中 \( T = T_1 + T_2 + \cdots \)。
动态关联通过双激发算符 \( T_2 \) 主导。
2. 静态关联处理方法#
多参考态展开(CASSCF):
选择活性空间 \( \text{Span}\{\phi_p\}_{p=1}^m \),构建多参考波函数:\[ |\Psi_{\text{CAS}}\rangle = \sum_{I} c_I |\Phi_I^{\text{active}}\rangle \]其中 \( |\Phi_I^{\text{active}}\rangle \) 为活性空间内的全组态展开。
密度矩阵重整化群(DMRG):
通过矩阵乘积态(MPS)参数化多体波函数:- \[ |\Psi_{\text{DMRG}}\rangle = \sum_{\{n_i\}} A^{n_1} A^{n_2} \cdots A^{n_N} |n_1 n_2 \cdots n_N\rangle \]
适用于强关联一维体系。
五、统一分类框架#
特征 |
动态关联 |
静态关联 |
|---|---|---|
能量尺度 |
激发态与参考态能量差大 |
多个参考态能量接近 |
波函数结构 |
单参考基态, 系数 \(c_0\) 主导 |
单激发态系数 \(c_i\) 接近基态 |
哈密顿矩阵 |
对角优势 |
分块对角化(强非对角耦合) |
处理方法 |
微扰理论、CC |
CASSCF、DMRG |
物理实例 |
H₂基态、小分子振动能级 |
O₂解离、过渡金属配合物 |
六、Python示例#
使用以下例子,可以更加真切的看到因为各种效应导致的能量差别:
基组截断#
1import numpy as np
2import matplotlib.pyplot as plt
3from pyscf import gto, scf, dft
4
5# 定义键长范围和基组
6bond_lengths = np.linspace(0.5, 3.0, 20)
7basis_sets = ['sto-3g', '6-31g', 'cc-pvdz', 'cc-pvtz']
8
9plt.figure(figsize=(10,6),dpi=300)
10for basis in basis_sets:
11 hf_energies = []
12 dft_energies = []
13
14 for r in bond_lengths:
15 # 构建分子
16 mol = gto.M(
17 atom=f'H 0 0 0; H 0 0 {r}',
18 basis=basis,
19 spin=0,
20 verbose=0
21 )
22
23 # HF计算
24 mf_hf = scf.RHF(mol).run()
25 hf_energies.append(mf_hf.e_tot)
26
27 # DFT计算 (LDA)
28 mf_dft = dft.RKS(mol, xc='lda,vwn').run()
29 dft_energies.append(mf_dft.e_tot)
30
31 # 绘制曲线
32 plt.plot(bond_lengths, hf_energies, '--', label=f'Hartree-Fock/{basis.upper()}')
33 plt.plot(bond_lengths, dft_energies, '-', label=f'DFT-LDA/{basis.upper()}')
34
35plt.title('several BASIS-SET in HF&DFT calc')
36plt.xlabel('bond length (Å)')
37plt.ylabel('energy (Ha)')
38plt.legend(bbox_to_anchor=(1.05, 1), loc='upper left')
39plt.grid(True)
40plt.show()
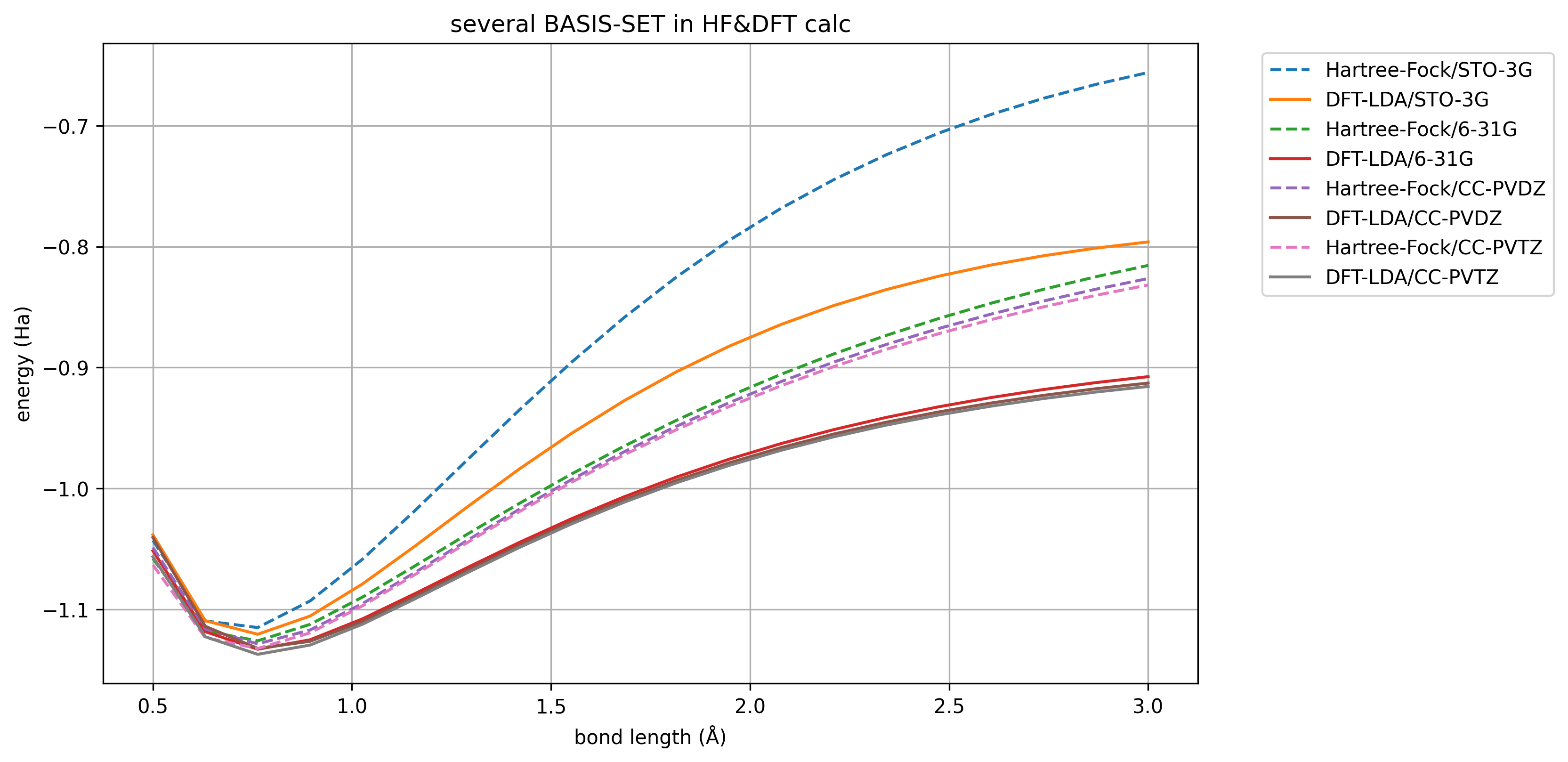
图 1.4.1 \(H_2\) 解离曲线,在不同基组下。#
这里我们模拟了不同基组下,HF 和 DFT 的结果差异。虚线是 HF 的结果,实线是 DFT. 可以看到,基组越大能量越低,但是到达一定程度也就够了。
交换能#
一个有趣的事实
这里可以看到常见泛函"B3LYP"的定义式,来自于若干个基础的泛函。从这里也可以感受到 DFT 方法简洁而高效。
1# 1. 仅Hartree项(无交换)
2class NoExchangeHF(scf.hf.RHF):
3 def get_veff(self, mol=None, dm=None, dm_last=0, vhf_last=0, hermi=1):
4 vj, _ = self.get_jk(mol, dm)
5 return vj # 只保留库伦项
6
7# Note B3LYP functional mf.xc = 'HF*0.2 + .08*LDA + .72*B88, .81*LYP + .19*VWN'
8# 2. 仅LDA交换(无关联)
9class LDA_X(dft.rks.RKS):
10 def __init__(self, mol):
11 super().__init__(mol)
12 self.xc = '0.0*LDA + 0.0*HF, 0.0*VWN' # 只使用交换项
13
14# 计算不同方法
15methods = {
16 'HF-NoX': [],
17 'DFT-NoXC': [],
18 'HF': [],
19 'DFT': []
20}
21
22# energies = {name: [] for name in methods}
23for r in bond_lengths:
24 mol = gto.M(
25 atom=f'H 0 0 0; H 0 0 {r}',
26 basis='6-31g',
27 spin=0,
28 verbose=0
29 )
30
31 mf = NoExchangeHF(mol).run()
32 methods['HF-NoX'].append(mf.e_tot)
33
34 mdft = LDA_X(mol).run()
35 methods['DFT-NoXC'].append(mdft.e_tot)
36
37 mf = scf.RHF(mol).run()
38 methods['HF'].append(mf.e_tot)
39
40 mdft = dft.RKS(mol).run()
41 methods['DFT'].append(mdft.e_tot)
42
43# 绘图部分已略去
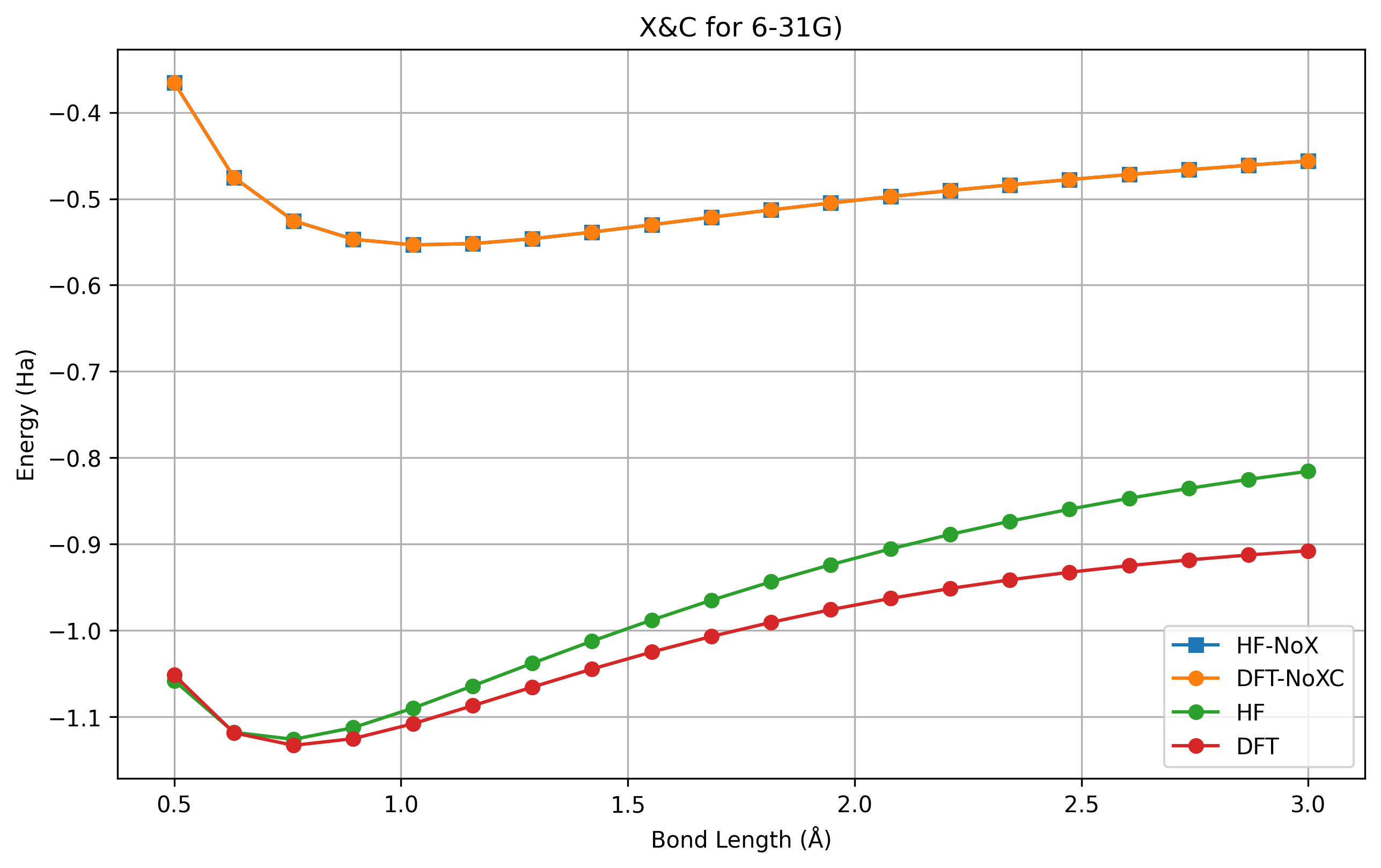
图 1.4.2 \(H_2\) 解离曲线,在交换能被关闭的情况下。#
这里,HF 和 DFT 在同一基组下,都被关闭了交换项。DFT 额外关闭了关联项,因为它不能单独关闭交换项。可以看到实际上如果不考虑交换关联,DFT 和 HF 的结果是一致,也就是直积态(单电子近似态)的能量。这是一个神奇的结果。而 HF 保留了交换项,能量大幅降低,DFT 考虑了交换和关联项,能量得以进一步降低。
关联能#
1from pyscf import mp, mcscf, fci
2
3plt.figure(figsize=(10,6),dpi=300)
4basis = 'sto3g'
5
6methods = {
7 'HF': [],
8 'MP2': [],
9 'CASSCF(2,2)': [],
10 'DFT-LDA': [],
11 'DFT-B3LYP': []
12}
13
14for r in bond_lengths:
15 mol = gto.M(
16 atom=f'H 0 0 0; H 0 0 {r}',
17 basis=basis,
18 spin=0,
19 verbose=0
20 )
21
22 # HF
23 mf = scf.RHF(mol).run()
24 methods['HF'].append(mf.e_tot)
25
26 # MP2
27 pt = mp.MP2(mf).run()
28 methods['MP2'].append(pt.e_tot)
29
30 # FCI
31 mc = mcscf.CASSCF(mf, 2, 2).run()
32 methods['CASSCF(2,2)'].append(mc.e_tot)
33
34 # DFT-LDA
35 mf_lda = dft.RKS(mol, xc='lda,vwn').run()
36 methods['DFT-LDA'].append(mf_lda.e_tot)
37
38 # DFT-B3LYP
39 mf_b3lyp = dft.RKS(mol, xc='b3lyp').run()
40 methods['DFT-B3LYP'].append(mf_b3lyp.e_tot)
41
42# 以HF为基准做相对能量
43ref_energy = min(methods['HF'])
44for method in methods:
45 plt.plot(bond_lengths, np.array(methods[method]) - ref_energy,
46 label=method, marker='o')
47
48# 绘图部分已略去
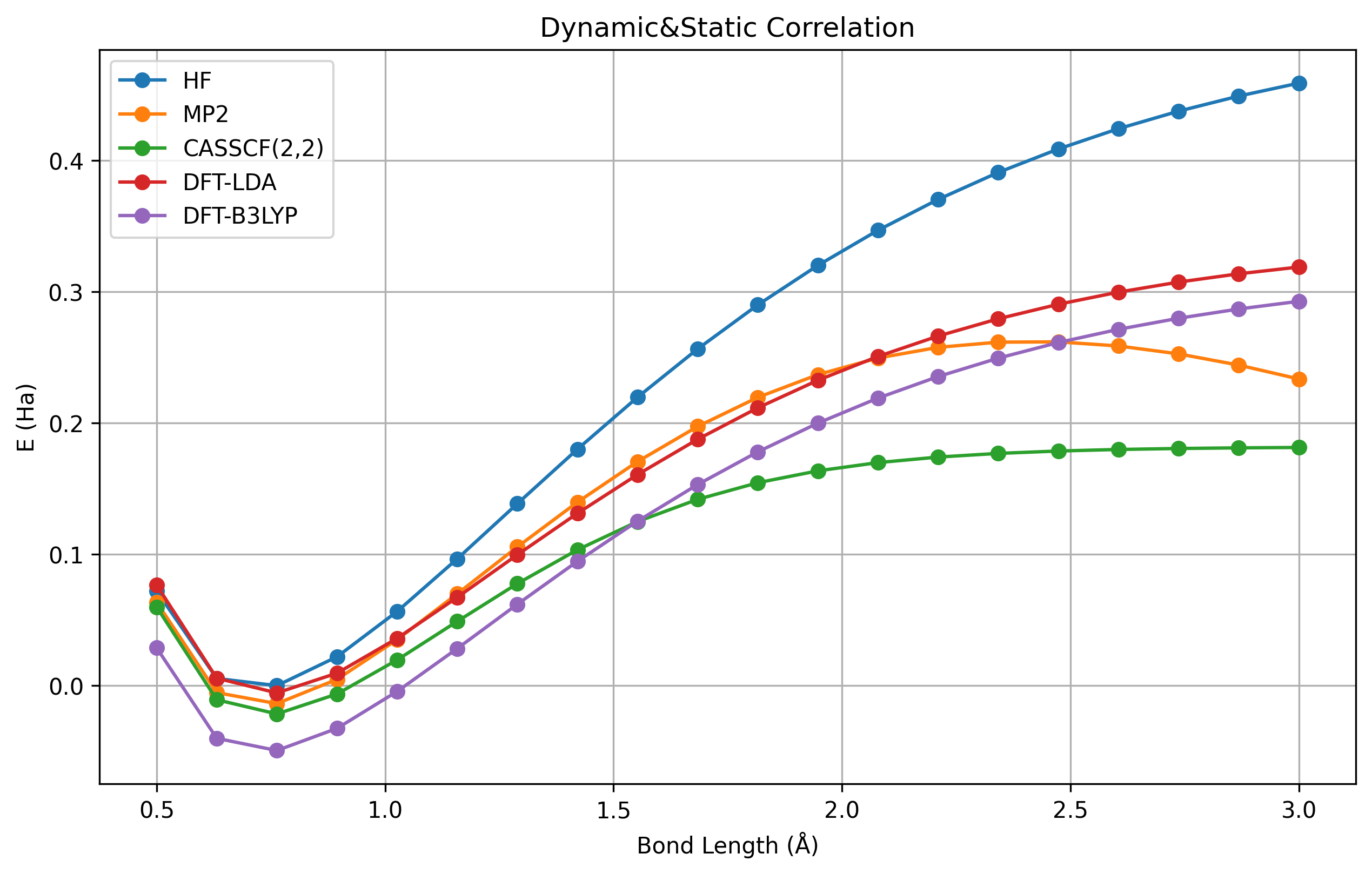
图 1.4.3 \(H_2\) 解离曲线,在不同的方法下,可以显示其关联效应。#
这里,HF 和 DFT 以及其升级方法,在同一基组下进行了计算。其中平衡位置 (\( \sim 0.74 \mathring {\mathrm A} \)) 主要是动态关联,HF 和 LDA 处理的都不好,但 MP2 一定程度上处理了这个问题。多参考,或者说静态关联区域在 \(1.5\sim 3 \mathring {\mathrm A} \) 左右,这里就需要 CASSCF 来处理,B3LYP处理的也不是很好。另一方面要注意 DFT 方法不是严格变分(因此猜测出来的泛函不来自于电子薛定谔方程)的,因此在平衡位置能量低于 CASSCF,这是错误的。